

Intel MPI
Displaying Intel-MPI Debug Information
The Detailed Information can be found at Displaying MPI Debug Information
The I_MPI_DEBUG environment variable provides a convenient way to get detailed information about an MPI application at runtime. You can set the variable value from 0 (the default value) to 1000. The higher the value, the more debug information you get.
High values of I_MPI_DEBUG can output a lot of information and significantly reduce performance of your application. A value of I_MPI_DEBUG=5 is generally a good starting point, which provides sufficient information to find common errors.
Displaying MPI Debug Information
To redirect the debug information output from stdout to stderr or a text file, use the I_MPI_DEBUG_OUTPUT environment variable
$ mpirun -genv I_MPI_DEBUG=5 -genv I_MPI_DEBUG_OUTPUT=debug_output.txt -n 32 ./mpi_programI_MPI_DEBUG Arguments
| <level> | Indicate the level of debug information provided. |
| 0 | Output no debugging information. This is the default value. |
| 1 | Output libfabric* version and provider. |
| 2 | Output information about the tuning file used. |
| 3 | Output effective MPI rank, pid and node mapping table. |
| 4 | Output process pinning information. |
| 5 | Output environment variables specific to the Intel® MPI Library. |
| > 5 | Add extra levels of debug information. |
| <flags> | Comma-separated list of debug flags |
| pid | Show process id for each debug message. |
| tid | Show thread id for each debug message for multithreaded library. |
| time | Show time for each debug message. |
| datetime | Show time and date for each debug message. |
| host | Show host name for each debug message. |
| level | Show level for each debug message. |
| scope | Show scope for each debug message. |
| line | Show source line number for each debug message. |
| file | Show source file name for each debug message. |
| nofunc | Do not show routine name. |
| norank | Do not show rank. |
| nousrwarn | Suppress warnings for improper use case (for example, incompatible combination of controls). |
| flock | Synchronize debug output from different process or threads. |
| nobuf | Do not use buffered I/O for debug output. |
References:
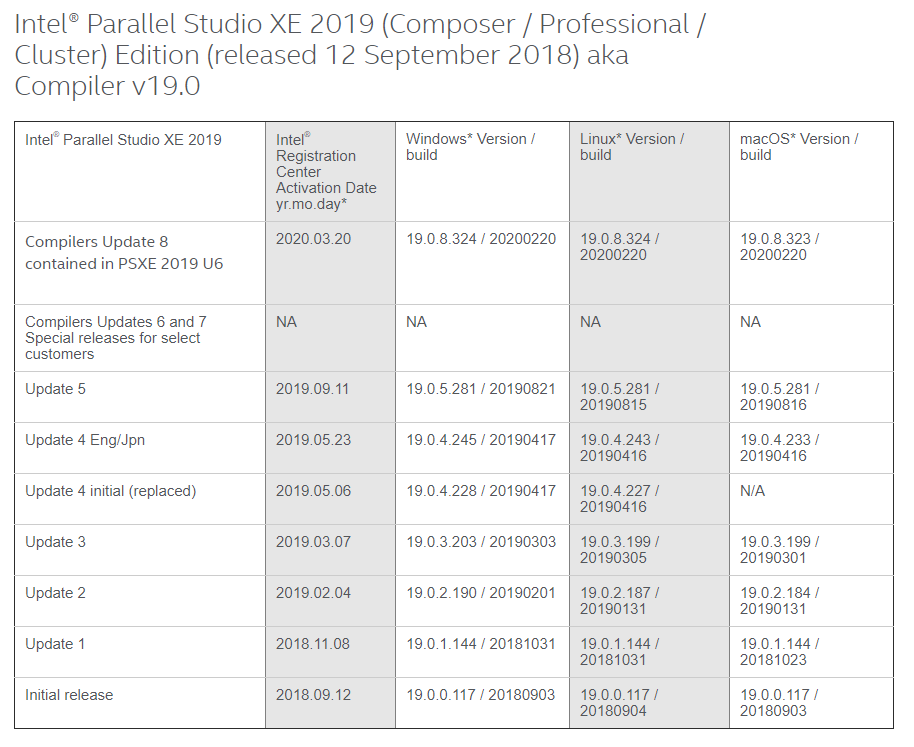
Intel Compilers Edition Update Version Mapping
This Website is important to get the right mapping for the Version Running Mapping from Intel. This is Good Information
Just took a snapshot of Intel 2018 to 2020


Intel MPI Library Over Libfabric*
Taken from Intel Performance Libraries, Intel® MPI Library Over Libfabric*

What is Libfabric?
Libfabric is a low-level communication abstraction for high-performance networks. It hides most transport and hardware implementation details from middleware and applications to provide high-performance portability between diverse fabrics.
Using the Intel MPI Library Distribution of Libfabric
By default, mpivars.sh sets the environment to the version of libfabric shipped with the Intel MPI Library. To disable this, use the I_MPI_OFI_LIBRARY_INTERNAL environment variable or -ofi_internal (by default ofi_internal=1)
# source /usr/local/intel/2018u3/impi/2018.3.222/bin64/mpivars.sh -ofi_internal=1
# I_MPI_DEBUG=4 mpirun -n 1 IMB-MPI1 barrier[0] MPI startup(): libfabric version: 1.7.2a-impi
[0] MPI startup(): libfabric provider: verbs;ofi_rxm
[0] MPI startup(): Rank Pid Node name Pin cpu
[0] MPI startup(): 0 130358 hpc-n1 {0,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,
30,31}
#------------------------------------------------------------
# Intel(R) MPI Benchmarks 2019 Update 4, MPI-1 part
#------------------------------------------------------------
# Date : Thu May 20 12:57:03 2021
# Machine : x86_64
# System : Linux
# Release : 3.10.0-693.el7.x86_64
# Version : #1 SMP Tue Aug 22 21:09:27 UTC 2017
# MPI Version : 3.1
# MPI Thread Environment:
# Calling sequence was:
# IMB-MPI1 barrier
# Minimum message length in bytes: 0
# Maximum message length in bytes: 4194304
#
# MPI_Datatype : MPI_BYTE
# MPI_Datatype for reductions : MPI_FLOAT
# MPI_Op : MPI_SUM
#
#
# List of Benchmarks to run:
# Barrier
#---------------------------------------------------
# Benchmarking Barrier
# #processes = 1
#---------------------------------------------------
#repetitions t_min[usec] t_max[usec] t_avg[usec]
1000 0.08 0.08 0.08
# All processes entering MPI_Finalize
Changing the -ofi_internal=0
# source /usr/local/intel/2018u3/impi/2018.3.222/bin64/mpivars.sh -ofi_internal=0
# I_MPI_DEBUG=4 mpirun -n 1 IMB-MPI1 barrier[0] MPI startup(): libfabric version: 1.1.0-impi
[0] MPI startup(): libfabric provider: mlx
.....
.....Common OFI Controls
To select the OFI provider from the libfabric library, you can use definte the name of the OFI Provider to load
export I_MPI_OFI_PROVIDER=tcpLogging Interfaces
FI_LOG_LEVEL=<level> controls the amount of logging data that is output. The following log levels are defined:
- Warn: Warn is the least verbose setting and is intended for reporting errors or warnings.
- Trace: Trace is more verbose and is meant to include non-detailed output helpful for tracing program execution.
- Info: Info is high traffic and meant for detailed output.
- Debug: Debug is high traffic and is likely to impact application performance. Debug output is only available if the library has been compiled with debugging enabled.
References:
Compiling Quantum ESPRESSO-6.5.0 with Intel MPI 2018 on CentOS 7
Step 1: Download Quantum ESPRESSO 6.5.0 from Quantum ESPRESSO Download Site or git-clone QE
% git clone https://gitlab.com/QEF/q-e.gitStep 2: Remember to source the Intel Compilers and indicate MKLROOT in your .bashrc
source /usr/local/intel/2018u3/mkl/bin/mklvars.sh intel64
source /usr/local/intel/2018u3/parallel_studio_xe_2018/bin/psxevars.sh intel64
source /usr/local/intel/2018u3/compilers_and_libraries/linux/bin/compilervars.sh intel64
source /usr/local/intel/2018u3/impi/2018.3.222/bin64/mpivars.sh intel64Step 3: Make a file call setup.sh and copy the contents inside.
export F90=mpiifort
export F77=mpiifort
export MPIF90=mpiifort
export CC=mpiicc
export CPP="icc -E"
export CFLAGS=$FCFLAGS
export AR=xiar
export BLAS_LIBS=""
export LAPACK_LIBS="-lmkl_blacs_intelmpi_lp64"
export SCALAPACK_LIBS="-lmkl_scalapack_lp64 -lmkl_blacs_intelmpi_lp64"
export FFT_LIBS="-L$MKLROOT/intel64"
./configure --enable-parallel --enable-openmp --enable-shared --with-scalapack=intel --prefix=/usr/local/espresso-6.5.0% ./setup.sh% make all -j 16
% make installChecking of availabilities of Libraries
% ./configure --prefix=/usr/local/espresso-6.5.0 --enable-parallel --enable-openmp --enable-shared --with-scalapack=intel | tee Configure.outChecking Configure.Out, there are some missing libraries which you have to fix.
..... ..... ESPRESSO can take advantage of several optimized numerical libraries (essl, fftw, mkl...). This configure script attempts to find them, but may fail if they have been installed in non-standard locations. If a required library is not found, the local copy will be compiled. The following libraries have been found: BLAS_LIBS= -lblas LAPACK_LIBS=-L/usr/local/lib -llapack -lblas FFT_LIBS= ..... .....
References:
Compiling OpenFOAM-5.0 with Intel-MPI
Minimum Requirements version
- gcc: 4.8.5
- cmake: 3.3 (required for ParaView and CGAL build)
- boost: 1.48 (required for CGAL build)
- fftw: 3.3.7 (optional – required for FFT-related functionality)
- Qt: 4.8 (optional – required for ParaView build)
I’m using Intel-16.0.4 and Intel-MPI-5.1.3.258
Step 1a: Download and Unpacking Sources
# wget -O - http://dl.openfoam.org/source/5-0 | tar xvz # wget -O - http://dl.openfoam.org/third-party/5-0 | tar xvz
Step 1b: Rename the Directory
# mv OpenFOAM-5.x-version-5.0 OpenFOAM-5.0 # mv ThirdParty-5.x-version-5.0 ThirdParty-5.0
Step 2: Initiate Intel and Intel-MPI Environment and source OpenFOAM-5.0 bashrc
source /usr/local/intel/bin/compilervars.sh intel64 source /usr/local/intel/parallel_studio_xe_2016.4.072/bin/psxevars.sh intel64 source /usr/local/intel/impi/5.1.3.258/bin64/mpivars.sh intel64 source /usr/local/intel/mkl/bin/mklvars.sh intel64
source /usr/local/OpenFOAM/OpenFOAM-5.0/etc/bashrc export MPI_ROOT=/usr/local/intel/impi/5.1.3.258/intel64
Step 3: Make sure your CentOS-7 Environment have the following base packages
# yum install gcc-c++ gcc-gfortran gmp flex flex-devel boost zlib zlib-devel qt4 qt4-devel
Step 4: Edit the OpenFOAM internal bashrc
# vim /usr/local/OpenFOAM/OpenFOAM-5.0/etc/bashrc
Line 35,36
export WM_PROJECT=OpenFOAM export WM_PROJECT_VERSION=5.0
Line 45
FOAM_INST_DIR=/usr/local/$WM_PROJECT
Line 60
export WM_COMPILER_TYPE=system
Line 65
export WM_COMPILER=Icc
Line 88
export WM_MPLIB=INTELMPI
Step 5: Compile OpenFOAM
# ./Allwmake -update -j
Intel MPI Parameter to consider for Performance
Selection of best available communication fabrics
Suggestion 1:
| I_MPI_DEVICE | I_MPI_FABRICS | Description |
|---|---|---|
| sock | tcp | TCP/IP-enable network fabrics, such as Ethernet and Infiniband* (through IPoIB*) |
| shm | shm | Shared-memory only |
| ssm | shm:tcp | Shared-memory + TCP/IP |
| rdma | dapl | DAPL-capable network fabrics, such as Infiniband*, iWarp*, Dolphon*, and XPMEM* (through DAPK*) |
| rdssm | shm:dapl | Shared-Memory + DAPL + sockers |
| ofa | OFA-capable network fabrics including Infiniband* (through OFED* verbs) | |
| tmi | TMI-capable network fabrics including Qlogic*, Myrinet* (through Tag Matching Interface) |
Suggestion 2:
| I_MPI_DAPL_UD | Values | Description |
|---|---|---|
| enable |
|
Suggestion 3:
| I_MPI_PERHOST | Values | Remarks |
|---|---|---|
| 1 | Make round-robin distirbution (Default value) | |
| all | Maps processes to all logical CPUs on a node | |
| allcores | Maps processes to all physical CPUs on a node |
Suggestion 4:
| I_MPI_SHM_BYPASS | Values | Remarks |
|---|---|---|
| disable | Set I_MPI_SHM_BYPASS* to ‘enable’ to turn on RDMA data exchange within single node that may outperform regular shared memory exchange. This is normally happens for large (350kb+) messages. |
Suggestion 5:
| I_MPI_ADJUST_ALLREDUCE | Values | Remarks |
|---|---|---|
| recursive doubling algorithm | 1 | |
| Rabenseifner’s algorithm | 2 | |
| Reduce + Bcast | 3 | |
| Topology aware Reduce + Bcast algorithm | 4 | |
| Binomial gather + scatter algorithm | 5 | |
| Topology Aware Binomial Gather + scatter algorithm | 6 | |
| Ring Algorithm | 7 |
Suggesion 6:
| I_MPI_WAIT_MODE | Values | Remarks |
|---|---|---|
| 1 | Set I_MPI_WAIT_MODE ‘to enable’ to try wait mode of the progress engine. The processes that waits for receiving that waits for receiving messages without polling of the fabrics(d) can save CPU time.
Apply wait mode to oversubscribe jobs |
References:
Using Intel IMB-MPI1 to check Fabrics and expected performances
In your .bashrc, do source the
source /usr/local/intel_2015/parallel_studio_xe_2015/bin/psxevars.sh intel64 source /usr/local/intel_2015/impi/5.0.3.049/bin64/mpivars.sh intel64 source /usr/local/intel_2015/composerxe/bin/compilervars.sh intel64 source /usr/local/intel_2015/mkl/bin/mklvars.sh intel64 MKLROOT=/usr/local/intel_2015/mkl
To simulate 3 workloads pingpong, sendrecv, and exchange with IMB-MPT1
$ mpirun -r ssh -RDMA -n 512 -env I_MPI_DEBUG 5 IMB-MPT1
Compiling with NWChem-6.6 with Intel MPI-5.0.3
Here is a write-up of my computing platform and applications:
- NWChem 6.6 (Oct 2015)
- Intel Compilers 2015 XE (version 15.0.6)
- Intel MPI (5.0.3)
- Intel MKL (11.2.4)
- Infiniband Inteconnect (OFED 1.5.3)
- CentOS 6.3 (x86_64)
Step 1: First thing first, source the intel components setting from
source /usr/local/intel_2015/parallel_studio_xe_2015/bin/psxevars.sh intel64 source /usr/local/intel_2015/impi/5.0.3.049/bin64/mpivars.sh intel64 source /usr/local/intel_2015/composerxe/bin/compilervars.sh intel64 source /usr/local/intel_2015/mkl/bin/mklvars.sh intel64
Step 2: Assuming you are done, you may want to download the NWChem 6.6 from NWChem Website. You may also want to take a look at instruction set for Compiling NWChem.
I have less problem running NWCHEM when the installation and the compiling directories are the same. If you recompile, do untar from source. Somehow the “make clean” does not clean the directories properly.
# tar -zxvf Nwchem-6.6.revision27746-src.2015-10-20.tar.gz # cd nwchem-6.6
Step 3: Apply All the Patches for the 27746 revision of NWChem 6.6
cd $NWCHEM_TOP wget http://www.nwchem-sw.org/download.php?f=Xccvs98.patch.gz gzip -d Xccvs98.patch.gz patch -p0 < Xccvs98.patch
Here is my nwchem_script_Feb2017.sh. For more details information, see Compiling NWChem for details on some of the parameters
export TCGRSH=/usr/bin/ssh export NWCHEM_TOP=/home/melvin/Downloads/nwchem-6.6 export NWCHEM_TARGET=LINUX64 export NWCHEM_MODULES=all export LARGE_FILES=TRUE export ARMCI_NETWORK=OPENIB export IB_INCLUDE=/usr/include export IB_LIB=/usr/lib64 export IB_LIB_NAME="-libumad -libverbs -lpthread" export MSG_COMMS=MPI export USE_MPI=y export USE_MPIF=y export USE_MPIF4=y export MPI_LOC=/usr/local/RH6_apps/intel_2015/impi_5.0.3/intel64 export MPI_LIB=$MPI_LOC/lib export MPI_INCLUDE=$MPI_LOC/include export LIBMPI="-lmpigf -lmpigi -lmpi_ilp64 -lmpi" export FC=ifort export CC=icc export MKLLIB=/usr/local/RH6_apps/intel_2015/mkl/lib/intel64 export MKLINC=/usr/local/RH6_apps/intel_2015/mkl/include export PYTHONHOME=/usr export PYTHONVERSION=2.6 export USE_PYTHON64=y export PYTHONLIBTYPE=so sed -i 's/libpython$(PYTHONVERSION).a/libpython$(PYTHONVERSION).$(PYTHONLIBTYPE)/g' config/makefile.h export HAS_BLAS=yes export BLAS_SIZE=8 export BLASOPT="-L$MKLLIB -lmkl_intel_ilp64 -lmkl_core -lmkl_sequential -lpthread -lm" export LAPACK_LIBS="-L$MKLLIB -lmkl_intel_ilp64 -lmkl_core -lmkl_sequential -lpthread -lm" export SCALAPACK_SIZE=8 export SCALAPACK="-L$MKLLIB -lmkl_scalapack_ilp64 -lmkl_intel_ilp64 -lmkl_core -lmkl_sequential -lmkl_blacs_intelmpi_ilp64 -lpthread -lm" export USE_64TO32=y echo "cd $NWCHEM_TOP/src" cd $NWCHEM_TOP/src echo "BEGIN --- make realclean " make realclean echo "END --- make realclean " echo "BEGIN --- make nwchem_config " make nwchem_config echo "END --- make nwchem_config " echo "BEGIN --- make" make CC=icc FC=ifort FOPTIMIZE=-O3 -j4 echo "END --- make " cd $NWCHEM_TOP/src/util make CC=icc FC=ifort FOPTIMIZE=-O3 version make CC=icc FC=ifort FOPTIMIZE=-O3 cd $NWCHEM_TOP/src make CC=icc FC=ifort FOPTIMIZE=-O3 link
General Site Installation
Determine the local storage path for the install files. (e.g., /usr/local/NWChem).
Make directories
# mkdir /usr/local/nwchem-6.6 # mkdir /usr/local/nwchem-6.6/bin # mkdir /usr/local/nwchem-6.6/data
Copy binary
# cp $NWCHEM_TOP/bin/ /usr/local/nwchem-6.6/bin # cd /usr/local/nwchem-6.6/bin # chmod 755 nwchem
Copy libraries
# cd $NWCHEM_TOP/src/basis # cp -r libraries /usr/local/nwchem-6.6/data # cd $NWCHEM_TOP/src/ # cp -r data /usr/local/nwchem-6.6 # cd $NWCHEM_TOP/src/nwpw # cp -r libraryps /usr/local/nwchem-6.6/data
The Final Lap (From Compiling NWChem)
Each user will need a .nwchemrc file to point to these default data files. A global one could be put in /usr/local/nwchem-6.6/data and a symbolic link made in each users $HOME directory is probably the best plan for new installs. Users would have to issue the following command prior to using NWChem: ln -s /usr/local/nwchem-6.6/data/default.nwchemrc $HOME/.nwchemrc
Contents of the default.nwchemrc file based on the above information should be:
nwchem_basis_library /usr/local/nwchem-6.6/data/libraries/ nwchem_nwpw_library /usr/local/nwchem-6.6/data/libraryps/ ffield amber amber_1 /usr/local/nwchem-6.6/data/amber_s/ amber_2 /usr/local/nwchem-6.6/data/amber_q/ amber_3 /usr/local/nwchem-6.6/data/amber_x/ amber_4 /usr/local/nwchem-6.6/data/amber_u/ spce /usr/local/nwchem-6.6/data/solvents/spce.rst charmm_s /usr/local/nwchem-6.6/data/charmm_s/ charmm_x /usr/local/nwchem-6.6/data/charmm_x/
References:
Compiling VASP-5.2.12 with Intel MPI-5.0.3
Vienna Ab initio Simulation Package (VASP) is a computer program for atomic scale materials modelling, e.g. electronic structure calculations and quantum-mechanical molecular dynamics, from first principles.
A. Prerequisites
To Compile VASP-5.2.12, I used
- Intel Compiler 15.0.6
- Intel MPI 5.0.3
- Maths Kernel Library 11.2.4
B. Compiling VASP Libraries
Assuming you have unpacked the VASP files. Here is may make file
.SUFFIXES: .inc .f .F #----------------------------------------------------------------------- # Makefile for LINUX NAG f90 #----------------------------------------------------------------------- # fortran compiler FC=ifort # C-preprocessor #CPP = /usr/lib/gcc-lib/i486-linux/2.7.2/cpp -P -C $*.F >$*.f CPP = gcc -E -P -C -DLONGCHAR $*.F >$*.f CFLAGS = -O FFLAGS = -Os -FI FREE = -FR DOBJ = preclib.o timing_.o derrf_.o dclock_.o diolib.o dlexlib.o drdatab.o #----------------------------------------------------------------------- # general rules #----------------------------------------------------------------------- libdmy.a: $(DOBJ) linpack_double.o -rm libdmy.a ar vq libdmy.a $(DOBJ) linpack_double.o: linpack_double.f $(FC) $(FFLAGS) $(NOFREE) -c linpack_double.f # files which do not require autodouble lapack_double.o: lapack_double.f $(FC) $(FFLAGS) $(NOFREE) -c lapack_double.f lapack_single.o: lapack_single.f $(FC) $(FFLAGS) $(NOFREE) -c lapack_single.f #lapack_cray.o: lapack_cray.f # $(FC) $(FFLAGS) $(NOFREE) -c lapack_cray.f .c.o: $(CC) $(CFLAGS) -c $*.c .F.o: $(CPP) $(FC) $(FFLAGS) $(FREE) $(INCS) -c $*.f .F.f: $(CPP) .f.o: $(FC) $(FFLAGS) $(FREE) $(INCS) -c $*.f
C. Compiling VASP
1. Copy the Makefile from makefile.linux_ifc_P4 in the vasp software.
# cp makefile.linux_ifc_P4 Makefile
2. Edit the Makefile
FC
#----------------------------------------------------------------------- # fortran compiler and linker #----------------------------------------------------------------------- FC=mpiifort # fortran linker FCL=$(FC)
CPP
CPP = $(CPP_) -DMPI -DHOST=\"LinuxIFC\" -DIFC \
-DCACHE_SIZE=32000 -DPGF90 -Davoidalloc -DNGZhalf \
-DMPI_BLOCK=64000 -Duse_collective -DscaLAPACK
FFLAGS
MKLROOT=/usr/local/RH6_apps/intel_2015/mkl MKL_PATH=$(MKLROOT)/lib/intel64 FFLAGS = -FR -names lowercase -assume byterecl -I$(MKLROOT)/include/fftw
OFLAG
#Haswell Architecture OFLAG=-O3 -xCORE-AVX2 #Sandy-Bridge Architecture OFLAG=-O3
The -xCORE-AVX2 is for Haswell Architecture
BLAS
BLAS= -mkl=cluster
-mkl=cluster is an Intel compiler flag that to include Intel MKL libraries, that will link with Intel MKL BLAS, LAPACK, FFT, ScaLAPACK functions that are used in VASP.
FFT3D
fftmpiw.o fftmpi_map.o fftw3d.o fft3dlib.o INCS = -I$(MKLROOT)/include/fftw
LAPACK and SCALAPACK
LAPACK= SCA=
Since the -mkl=cluster, includes MKL ScaLAPACK libraries also, it is not required to mentioned the ScaLAPACK libs. That include LAPACK
References:
- Building VASP* with Intel® MKL and Intel® Compilers
- (Intel Developer Zone)
